G_cluster Analysis Gromacs
Gmx clustize is used for cluster analysis in a narrow sense that is the aforementioned group of atoms is divided into different clusters. Distances between structures can be determined from a trajectory or read from an.
2
This allows subsequent analysis of the subtrajectories that could for example be the result of a cluster analysis.
G_cluster analysis gromacs. Asked 10th Feb 2015. There are several techniques available for finding information in GROMACS trajectory files. Use gmxdumpand redirect the shell output to a file and.
Gmx trjconv -f traj_compxtc -o clusterxtc -pbc cluster -dt 10 Select protein for clusering and protein for output. Xpm matrix file with the -dm option. To build GROMACS with OpenCL support enabled two components are required.
These are all structured as part of a single gmx wrapper binary and invoked with commands like gmx grompp. It is compatible with argparse. Gmx trjconv -f clusterxtc -o peptidepdb -fit rottrans When prompted to select groups select the backbone for the fitting and the protein for the output.
St rmsd middle rmsd cluster. How should I read a cluster output and what does it infer. If no -g is passed the program will read all the gro files inside the workdir.
We will first have a quick look at the peptide using VMD. Actually 12-HSA formed some clusters and i want to do cluster analysis of it. The option to write subtrajectories -sub based on the information obtained from cluster analysis has been removed from gmx trjconv and is now part of gmx extract-cluster gmx trjcat is better suited for concatenating multiple trajectory files.
Gmx gangle -n indexndx -g1 vectorplane -group1 group A -g2 zt0 -oav. The following formats are supported for input and output. GROMACS USER MANUAL Version 32 David van der Spoel Erik Lindahl Berk Hess Aldert R.
A -g GRO file. This program is compatible with GROMACS itp format for molecular topologies. Write your own C code using gromacssharetemplatetemplatecas a template.
Gmx cluster can perform generalized cluster analysis However the function realized by the program is only to perform cluster analysis on different conformations of proteins according to RMSD thereby. G_rms_467 -f finaltrr -s final. The GROMACS OpenCL on NVIDIA GPUs works but performance and other limitations make it less practical for details see the user guide.
RMS deviation after fitting or RMS deviation of atom-pair distances can. G_gyratecomputes the radius of gyration of a group of atoms and the radii of gyration about the x y and z axes as a function of time. I have simulated a protein for 100ns and the cluster analysis is performed.
Use the GROMACS trajectory analysis utilities. For more information run. Using gromos method for clustering Using RMSD cutoff 014 nm The RMSD ranges from 00637001 to 0388457 nm Average RMSD is 020783 Number of structures for matrix 1601 Energy of the matrix is 126831 nm Found 23 clusters Writing middle structure for each cluster to clusterspdb Counted 268 transitions in total max 30 between two specific clusters cl.
Is the structure provided by g_cluster GROMACS a real frame or an average. Peter Tieleman Alfons LTM. What does MD produce.
Analysis of MD trajectories in GROMACS David van der Spoel. Use g_trajto write a xvg fileand read that in an external program as above. The inputs are the following.
But i am ended up. Clustering algorithms can give an answer g_cluster implements a number of popular algorithms all of which have their own issues. Energy terms Et Coordinates xt Velocities vt Forces ft.
Mdrun is the only other binary that can be built. 29 rows g_cluster can cluster structures using several different methods. GROMACS includes many tools for preparing running and analyzing molecular dynamics simulations.
If you only had a single index group A in indexndx and you used g_sgangle -z or -one you can use. This assumes that the entries in the index file are frame numbers and dumps each group in the index file to a separate trajectory file. In the normal build it can be run with gmx mdrunDocumentation for these can be found at the respective sections below as well as on man.
For the distances you can use gmx distance to compute one or more distances as you want. I need to do cluster analysis of my MD simulation which has been done with gromacs-467. There are two programs involved in cluster analysis in GROMACS.
Select frames within a certain range of a quantity given in an xvg file. My commands are as following. The additional runtime-only dependency is the vendor-specific GPU driver for.
After invoking the command you are requested to select a group for which the calculation is to be performed. In gromacsanalysisplugins command_name in G_cluster command_name in G_tcaf CysAccessibility in gromacsanalysispluginscysaccessibility command_name in G_clustsize command_name in G_traj. I am thinking to use Gromacs for cluster analysis but it seemed Gromacs requires some kinds of input files that I totally have no idea.
The OpenCL headers and the wrapper library that acts as a client driver loader so-called ICD loader. Xtc trr gro g96 and pdb. Hi all Im trying to perform a cluster analysis on my MD simulation using g_cluster.
1first i measured RMSD by using following command. Im analysing a simulation of a. The various GROMACS analysis utilities can generate xvg filesThese are text files that have been specifically formated for direct use in GraceYou can however in all GROMACS analysis programs turn off the Grace specific codes by running the programs with the -xvg none optionThis circumvents problems with tools like gnuplot and Excel see below.
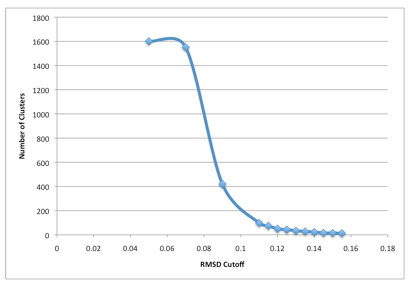
It must comply with gromacs GRO format. To choose a reasonable RMSD cut-off first we varied the C RMSD cut-off between 010 to 015 in steps of 001 and performed clustering analysis for each RMSD cut-off value. The xpm file is converted to eps.
The atoms are explicitly mass weighted. Van Buuren Emile Apol Pieter J. Both distances between centers of groups or individual atoms are supported using the new selection syntax.
GROMOS clustering algorithm described by Daura et al2 with a C RMSD cut-off was used to determine the structurally similar clusters.
Rmsd Based Clustering Tutorial Biochemcore 2018

How To Calculate Coordination Number In Gromacs

Computational And Experimental Analysis Of Drug Binding To The Influenza M2 Channel Sciencedirect
Posting Komentar untuk "G_cluster Analysis Gromacs"