G_cluster Gromacs 4.6
Peter Tieleman Alfons LTM. For example notice that cluster 1 the most-populated cluster has 805 members not all shown.
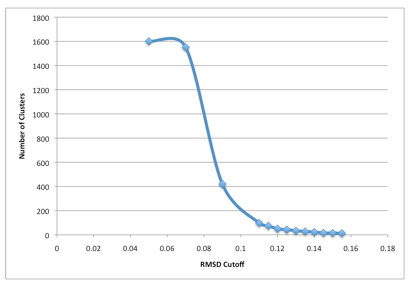
Rmsd Based Clustering Tutorial Biochemcore 2018
Cluster structures from Autodock runs.
G_cluster gromacs 4.6. If you have two big vesicles and you want to maintain their relative positions. With GROMACS 46 native GPU acceleration support is introduced. Use CUDA 50 or newer 70 for Maxwell.
Redirected from About GromacsRelease NotesVersions 46x. For GROMACS we require the compiler to support libstc version 461 or higher. Release notes for 467.
If compiling on a cluster head node make sure that GMX_CPU_ACCELERATION is appropriate for the compute nodes. Analyzes bundles of axes eg. SuperMUC is a cluster of 18432 Intel Xeon E-2680 Sandy Bridge CPUs with eight cores each operating at a clock rate of 27 GHz.
This report summarizes the results of GROMACS 46 benchmarks carried out during the SuperMUC Extreme Scaling Workshop at the Leibniz-Rechenzentrum LRZ in July 2013. It is free open source released under the GNU General Public License. By loading the gcc module.
Starting from version 46 GROMACS is released under the GNU Lesser General Public License. For Gromacs 407 there is a GPL tool that finds the optimal performance with PME on a given number of processors download g_tune_pme Unpack with tar -xvzf. The docked energy and free energy estimates are analysed and for each cluster the energy statistics are printed.
To select a particular libstdc library use. Results obtained with version 46 will in the majority of cases hold for version 50 since the performance of CPU and GPU compute kernels have not. Add a structure to a.
The native implementation of GPU support in GROMACS 46 and later is discussed on a separate page with more further details in the Acceleration and parallelization section and in the 46 manual section A6. Make sure your compiler supports OpenMP. MAN page from Fedora 21 gromacs-common-465-5fc21noarchrpm.
G_anadock analyses the results of an Autodock run and clusters the structures together based on distance or RMSD. GROMACS 46 and GPUs. Calculate size distributions of atomic clusters.
From GROMACS version 46. When velocities are present in your trajectory the temperature of the largest cluster will be printed in a separate xvg file assuming that the particles are free to move. The most compute-intensive part of simulations the non-bonded force calculation can be offloaded a GPU and carried out simultaneously with CPU calculations of bonded forces and PME eletrostatics.
1first i measured RMSD by using following command. Which has been done with gromacs-467. Various fixes to md-vv integrator back-ported from 46 branch includes fixes to aspects of the interaction of md-vv with various pressure coupling algorithms temperature coupling algorithms dispersion correction multiple threads preserving exact KE when nstcalcenergy is not a multiple of nstpcouple or nsttcouple 1116 1012 1000 1129.
29 rows g_cluster can cluster structures using several different methods. Use with a recent CUDA driver. Note that Gromacs starts numbering the frames with -1 while VMD starts numbering frames with 0.
GROMACS 463 Online Reference. The benchmarks have been carried out with the most recent version of GROMACS 46 available at the time of testing see 5th column of Table 2. GROMACS suite VERSION 465 1 Updated.
General Getting Started Flow Chart File Formats mdp options FAQ. The information below pertains only to the GPU support in GROMACS 45 series which is based on the OpenMM library. Gromacs-data_465-1build1_all NAME g_clustsize - calculate size distributions of atomic clusters VERSION 465 SYNOPSIS g_clustsize-f trajxtc-s topoltpr-n indexndx-o csizexpm-ow csizewxpm-nc nclustxvg-mc maxclustxvg-ac avclustxvg-hc histo-clustxvg-temp tempxvg-mcn maxclustndx-noh-noversion-nice int-b time-e time-dt time-tu enum-now-xvg enum-cut real-nomol.
The cluster is organized in. The most representative frame or the cluster centroid is the 1356 frame of the trajectory. But i am ended up getting following.
GROMACS USER MANUAL Version 32 David van der Spoel Erik Lindahl Berk Hess Aldert R. If you are using constraints please correct the temperature. Distances between structures can be determined from a trajectory or read from an.
Van Buuren Emile Apol Pieter J. From version 45 on g_tune_pme is part of the official Gromacs package. I am trying to compile Gromacs-462 for a GPU cluster with following command.
This is useful eg. Programs Options do_dssp editconf eneconv g_anadock g_anaeig g_analyze. -DGMX_STDLIB_CXX_FLAGS-gcc-namepathtogccbinary or make sure that the correct gcc version is first in path eg.
Load the gromacs module so that they are also in your path and they can be used without typing the full path. Here we report on the performance of GROMACS 46 on the SuperMUC cluster at the Leibniz Rechenzentrum in Garching. We carried out benchmarks with.
The separate option -clustercenter can be used to specify an approximate center for the cluster. RMS deviation after fitting or RMS deviation of atom-pair distances can be used to define the distance between structures. Release notes for 466.
Release notes for 457. Xpm matrix file with the -dm option. Actually 12-HSA formed some clusters and i want to do cluster analysis of it.
The central member of the cluster ie. The outpdb file produced by g_cluster contains one structure for each cluster identified using a specific cutoff. Release notes for fixes in the git repository made since 467 but not released 2.
The fastest CUDA versions with GROMACS are 55 65 and 75. CCicc FCifort F77ifort CXXicpc CMAKE_PREFIX_PATHexportintelcmklinclude. VERSION 463 Fri 5 Jul 2013.
VERSION 467 Fri 29 Aug 2014. There is also a poster describing g_tune_pme. My commands are as following.
Mon 2 Dec 2013 Index NAME g_cluster - clusters structures. GROMACS is one of the fastest and most popular molecular dynamics software packages available and can run on GPUs as well as CPUs. An alternative approach to this is to cluster the structures first using g_cluster and then sort the clusters on either lowest energy or average energy.
G_rms_467 -f finaltrr -s final.

Gromacs Cluster Analysis

How To Analyse A Cluster Xpm File Generated Using Gromacs

Gromacs 4 6 7 Online Reference
{kind=link}
Posting Komentar untuk "G_cluster Gromacs 4.6"